
PCR ---- is the future of molecular diagnostics
As mentioned in the previous article, molecular diagnostics is the fastest growing segment in the IVD industry. According to the technical classification, it can be divided into nucleic acid molecular hybridization technology, nucleic acid amplification technology, gene chip technology, gene sequencing technology. As one of the in vitro nucleic acid amplification technologies, PCR technology occupies more than 70% of the total amount of molecular diagnostic products due to its advantages of relatively low barriers, high degree of localization and relatively early layout of domestic enterprises. This article will give a brief introduction to the common techniques of the PCR family.
01 What is PCR?
Polymerase Chain Reaction (PCR) is a molecular biology technique for amplifying specific DNA fragments in vitro. Since its emergence, PCR technology has played a huge role in the field of biological science, molecular diagnosis, paternity test, forensic identification and criminal investigation, and is one of the most important technologies so far.

Figure 1 Development history of PCR instrument (Source: Lee NY., 2018[1])
Through evolution and innovation, PCR technology has been developed for three generations: ordinary PCR, quantitative real-time PCR(qPCR) and digital PCR(dPCR).
02 Principle of PCR
The target DNA fragment can be amplified more than one million times by PCR technology. The principle is that under the catalysis of DNA polymerase, the parent strand DNA is used as the template, and the specific primer is used as the extension starting point. Through denaturation, annealing, extension and other steps, the daughter strand DNA complementary to the parent strand template DNA is copied in vitro.
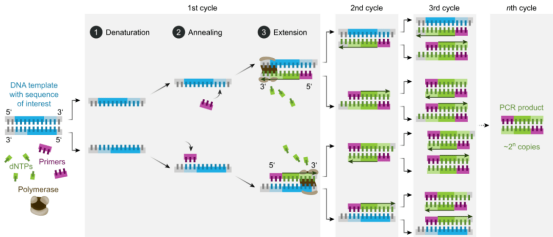
Figure 2 Basic reaction steps of PCR (Source: Network)
The standard PCR process is divided into three steps:
1. Denaturation: separation of template DNA double strand by high temperature. The hydrogen bond between the DNA double strands is broken at high temperatures (93-98 ℃) and dissociated into a single strand in order to bind to the primer.
2. Annealing: when the temperature has been lowered to about 55℃, the primers have been combined with single strand of template DNA according to the principle of base complementary pairing.
3. Can I have Extension? Then, the optimal reaction temperature of DNA polymerase was set to about 72℃. Under the action of Taq DNA polymerase, the DNA template and primer conjugate synthesized a new semi-retained replication chain complementary to the template DNA strand along the direction from phosphoric acid to five-carbon sugar (5'→3') according to the principle of base pairing and semi-retained replication. Repeating these three steps 25 to 35 times will increase the number of DNA fragments exponentially.
03 Types of technologyluorescence quantitative PCR
Quantitative Real-time PCR, qPCR
RT-qPCR (RT-QPCR) is an experimental method that uses RNA as the starting material, and a reverse transcription PCR technique combined with fluorescence quantitative technique. Compared with first-generation PCR, real-time fluorescent quantitative PCR has the advantages of second-generation PCR mainly in fluorescence, real-time and quantitative. A fluorescence probe was added into the PCR amplification reaction system, and the changes of the amount of amplified products in each cycle were monitored in real time through the collection of fluorescence signals. The standard curve and CT value were used for quantitative analysis of the tested samples. Commonly used fluorescent substances can be divided into TaqMan fluorescent probes, fluorescent dyes and molecular beacons.
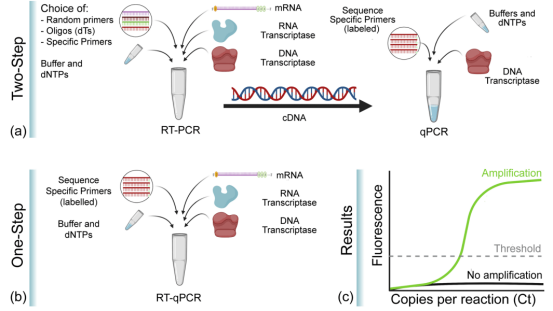
Figure 3 One-step and two-step RT-qPCR
(Source: Soler et al., 2020[2])
RT-qPCR has been used in a variety of molecular biology applications, including gene expression analysis, RNA interference validation, microarray validation, pathogen detection, gene testing, and disease research. RT-qPCR can be performed by a one - or two-step method. One step method combines reverse transcription with PCR amplification, so that reverse transcriptase and DNA polymerase complete the reaction in the same tube under the same buffer condition, this method only needs to use sequence specific primers. The reverse transcription and PCR amplification processes in the two-step method were performed in two tubes, using different optimized buffers, reaction conditions, and primer design strategies.
01
Fluorescent probe method (TaqMan technique)
The TaqMan probe is the earliest quantitative method and the most commonly used test method in clinical testing. In PCR amplification, a specific fluorescent probe was added along with a pair of primers. The probe is an oligonucleotide labeled with a fluorescene group (Reporter, R) at the 5' end and a quench group (Quencher, Q) at the 3' end. When the probe was intact, the fluorescence signal emitted by the reporting group was absorbed by the quenched group so that the fluorescence signal could not be detected. When PCR amplification (in the extension stage), the probe will be degraded by the 5'→3' exonuctant active enzyme of Taq enzyme, so that the reporter group and quenched group are separated, and the fluorescence of the reporter group emission base is no longer absorbed, so that the fluorescence signal can be received in the fluorescence monitoring system, that is, each amplified DNA will form a fluorescent molecule. The formation of PCR products is completely synchronized with the formation of fluorescent molecules. The more PCR products, the more fluorescence signals are accumulated, and the greater the fluorescence intensity.
This method has strong specificity, high sensitivity, and is suitable for multiple qPCR detection. Besides, there is no need for subsequent PCR processing, saving time and raw material cost. However, the hydrolysis of probe depends on the activity of Taq exonuclease, which is easy to be affected by reagent and enzyme properties when quantification, and it is difficult to quench thoroughly, with high background. It is not only necessary to synthesize different probes according to different sequences, but also difficult to judge the actual amplification characteristics of the detection results.
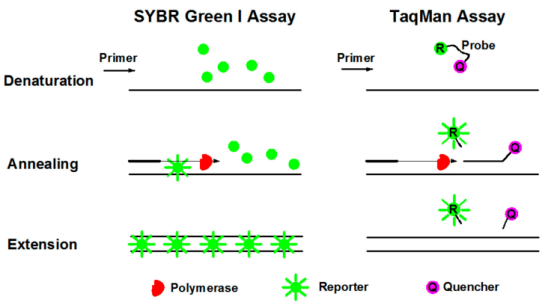
Figure 4. qpcr principle of fluorescent dye method (left) and fluorescent probe method (right)
(Source: Cao et al., 2020[3])
02
Fluorescent dye process (SYBR Green)
SYBR GreenⅠ is the most commonly used fluorescent dye in quantitative fluorescent PCR and binds to all double-stranded DNA. In the PCR reaction system, SYBR GreenⅠ was added, which would bind to the double-stranded DNA in the process and produce fluorescence signal. Therefore, the increase of fluorescence signal in the reaction was synchronized with the increase of PCR products, and the fluorescence intensity also increased with the increase of products. This method has the advantages of high sensitivity, relatively low price, no choice for DNA template, good universality and convenient use. However, since dyes bind to double-stranded DNA in a non-specific way, false positive results may be generated, so it is necessary to identify the specificity of amplified products by melting curve analysis. At the same time, it is necessary to optimize the reaction system to reduce non-specific amplification, which is not suitable for multiple qPCR detection.
03
Molecular beacon
It is a stem-ring double-labeled oligonucleotide probe that forms a hairpin structure of about 8 bases at the 5' and 3' ends. The nucleic acid sequences at both ends are complementary and paired, resulting in the close proximity of fluorescent groups and quenched groups without fluorescence.
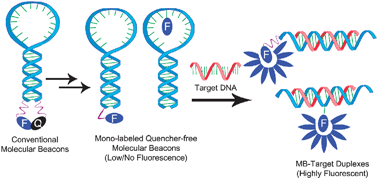
Figure 5 Working principle of molecular beacon
(Source: N. Venkatesan et al., 2008[4])
After the PCR product is generated, the middle part of the molecular beacon is paired with a specific DNA sequence during the annealing process, and the fluorescent gene is separated from the quenched gene to produce fluorescence.
Isothermal amplification technique
Isothermal Amplification Technology
Isothermal Amplification is a novel nucleic acid amplification technique developed in recent years based on Isothermal Amplification, which mainly includes Loop-mediated isothermal amplification, LAMP), Crossing Priming Amplification (CPA), Strand Displacement Amplification (Strand Displacement Amplification, SDA), Recombinase Polymerase Amplification (RPA), Recombinase polymerase Amplification (RPA), Nucleic Acid Sequence-based Amplification, NASBA), Rolling Circle Amplification (RCA) and Helicase-dependent Amplification (HDA). Although various isothermal amplification techniques adopt constant temperature amplification, the design of primers and amplification principles are quite different. Two of them will be briefly discussed.
01
Ring-mediated isothermal amplification
Loop-Mediated Isothermal Amplification (LAMP) was performed at 60~65 ℃, Four primers and a DNA polymerase of Bacillus stearothermophilus (BstDNA polymerase) with chain replacement function are required. The outer primer is similar to the PCR primer, while the inner primer contains two sequences. The process is as follows: (1) The inner primer binds the target gene and extends into double strand under the action of BstDNA polymerase. The outer primer binds to the 5 'end of the double-stranded DNA, forming a ring structure at one end. The other end goes through the same process to form a dumbbell-like structure with rings at both ends. (2) Single-stranded DNA with dumbbell structure has dual functions of template and primer, and can be extended immediately under the catalysis of Bst polymerase. (3) Inner primers can also bind to annular structures and extend under the action of enzymes.
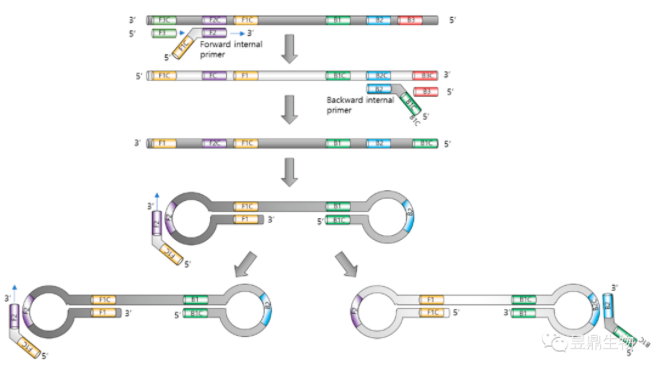
Figure 6 Principle of loop-mediated isothermal amplification
(Source: Park J-W et al.,2022[5])
02
Transcription based amplification
Transcription based Amplification includes Nucleic Acid Sequence-based Amplification (NASBA) and Transcription Mediated Amplification (TMA), Both are isothermal amplification techniques by RNA.
TMA is a pair of specific primers designed for the target sequence, in which the promoter primer has a promoter sequence recognized by T7 RNA polymerase. After combining with the target sequence, this primer carries out reverse transcription reaction under the action of reverse transcriptase to form RNA-DNA hybrid molecules. The RNase H activity of reverse transcriptase hydrolyzes RNA-DNA hybrid molecules to form single-stranded DNA containing promoter sequences recognized by T7 RNA polymerase. Primer 2 then binds to the single-stranded DNA and produces double-stranded DNA by reverse transcription. T7 RNA polymerase binds to the promoter and uses DNA as a template for transcription. 100 to 1000 copies of transcripts can be obtained from a molecular DNA template. These transcripts enter the reaction and serve as the starting template for TMA. In TMA reaction, the product RNA increases exponentially, and the target sequence can be amplified by about 1010 within 15 to 30 min [6]. Hybridization Protection Assay (HPA) was then used to assay the RNA products.

Figure 7. Principle of transcription-mediated amplification (Source: Network)
Digital PCR
Digital PCR,dPCR
Digital PCR, namely three-generation PCR, is another improvement of the original PCR. The copy number of the target sequence is calculated by the end point detection, and the absolute quantitative detection can be performed accurately without using internal parameters and standard curves. Endpoint detection is independent of Ct value (cyclic threshold), reduced by amplification efficiency, improved tolerance to PCR inhibitors, and has high accuracy and reproducibility. This method requires higher quality of template, the amount of template exceeds the amount of microsystem will lead to no quantitative, too little quantitative accuracy will be reduced. False positives can also occur in the presence of nonspecific amplification, and the instruments and reagents used are expensive.
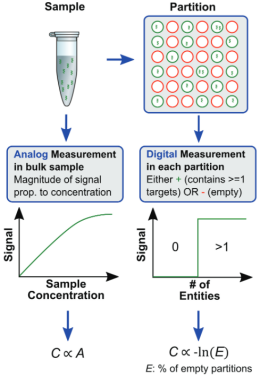
Figure 8 Principle of digital PCR (Source: Network)
Due to its characteristics of high sensitivity and high accuracy, digital PCR is not easy to be interfered by PCR reaction inhibitors, and absolute quantification in real sense can be achieved without standard substances, so it still becomes a research and application hotspot. According to the different forms of reaction units, the system can be divided into three categories: microflow, chip and microdrop.
04 Clinical significance
PCR technology is an indispensable tool that is widely used in the laboratory and in the clinic. It is often used to detect the presence of specific alleles, such as couples screening for genetic disease carriers during pregnancy or early pregnancy, which can directly diagnose the presence of diseases and mutations in developing embryos. It was noted in the literature that PCR was first used to detect a single gene mutation to diagnose sickle cell anemia [7].
In addition, PCR technology has dramatically changed the diagnostic potential of infectious diseases because it can be used to rapidly identify microorganisms that are difficult to culture or require long growth cycles [8]. Pathogens detected by routine PCR tests include mycobacterium tuberculosis, human immunodeficiency virus, herpes simplex virus, syphilis, and other pathogens. In addition, qPCR is not only used to detect the qualitative presence of microorganisms, but also can be used to quantify bacteria, fungi and viral load [9].
Thanks to the advent of PCR technology, the sensitivity to oncogene and tumor suppressor gene mutations has been increased at least 10,000 times, enabling earlier diagnosis of cancers such as leukemia. PCR also offers more refined and personalized treatment for cancer patients. In addition, PCR technology also has a great impact on the prenatal detection of various genetic diseases or clinicopathology [10].
In addition to the several PCRS described above, There are also Hot start PCR, Nested PCR, Touchdown PCR, Gene splicing by overlap extension PCR, gene splicing by Overlap extension PCR, SOE PCR), Inverse PCR (IPCR), Fast PCR, high GC content PCR (GC-rich PCR), Long Range PCR and other technologies. In summary, improved PCR protocols and improved DNA polymerase are designed to improve the results of PCR amplification. Although the basic concept of PCR has not changed, new PCR methods will continue to advance and simplify molecular biology research.
Reference literature
[1] Lee NY. A review on microscale polymerase chain reaction based methods in molecular diagnosis, and future prospects for the fabrication of fully integrated portable biomedical devices. Mikrochim Acta. 2018,185(6).
[2]N. Venkatesan, Y. Jun Seo and B. Hyean Kim,Quencher-free molecular beacons: a new strategy in fluorescence based nucleic acid analysis. Chem. Soc. Rev., 2008, 37, 648.
[3] Cao Y, Yu M, Dong G, et al. Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation. Molecules. 20,25(3).
[4] Soler M, Scholtz A, Zeto R, et al. Engineering photonics solutions for COVID-19. APL Photonics. 20,5(9).
[5] Park J-W. Principles and Applications of Loop-Mediated Isothermal Amplification to Point-of-Care Tests. Biosensors. 2022; 12 (10) : 857.
[6] Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. 1985. Biotechnology. 1992; 24:47 6-80.
[7] Hill C S. Molecular diagnostic testing for infectious diseases using TMA technology. Exp Rev Mol Diagn, 2001, 1 (4) : 445-455.
[8] Mackay IM. Real-time PCR in the microbiology laboratory. Clin Microbiol Infect. 2004 Mar; (3) : 190-212.
[9] Muldrew KL. Molecular diagnostics of infectious diseases. Curr Opin Pediatr. 2009 Feb; 21 (1) : 102-11.
[10] Ghannam, Mousa & Varacallo, Matthew. (2018). Biochemistry, Polymerase Chain Reaction (PCR).
